Jacobi Iteration
In this example we will get very close to a "real" application in
numerical fluid mechanics. We will solve the following Poisson in two
dimensions
L(u)=(dxx+dyy)u=0
on a unit square [0,1]x[0,1] with Dirichlet boundary conditions
u=sin(Pi*x)*exp(-Pi*y) for x=0, x=1, y=0, y=1
Using a Cartesian, uniform mesh and second-order accurate, central
finite-differences, the spatial operator is discretized as follows:
L(u)=(ui+1,j-2ui,j+ui-1,j)/dx2+(ui,j+1-2ui,j+ui,j-1)/dy2
for all 1<=i<=m, 1<=j<=n and - for simplicity - dx=dy. Note that
the boundaries correspond to the indices i=0,m+1 and j=0,n+1.
The Jacobi method for iteratively solving the above system can be
obtained as follows. First, modify the equations by introducing a
time-derivative:
dt u=L(u) -> (un+1-un)/dt=L(un)
We will iterate until dtu=0, which gives the solution to the
original problem. Substituting the discrete spatial operator and
taking the maximum stable "time step" gives the formula for the
Jacobi iteration:
un+1i,j=(ui+1,jn+ui-1,jn+ui,j+1n+ui,j-1n)/4
Note that the right-hand-side consists only of value at the previous
time level "n", and, therefore, the formula is explicit. This method
is not efficient for practical computations (converges slowly,
accumulates round-off) but serves its purpose as an example.
An implementation for a single processor is accessible
below.
How to approach the parallelization of the algorithm?
- Functional decomposition or domain decomposition?
-> there is nothing else to do but to execute the Jacobi formula on a
large amount of data many times. Therefore, we will certainly choose
a domain decomposition technique.
- How to decompose the domain/data?
Look at the algorithm. First observation: the formula is fully
explicit per time step. That means that once all required
previous values are known, all those values can be updated locally without need for communication. Second observation: the
right-hand-side for a given node (i,j) is a function of four
neighboring node values (i-1,j), (i+1,j), (i,j-1), (i,j+1). This
means that after each time step, we need to communicate a limited
amount of data. More specifically: for this type of 3-point
difference formulas (in each direction), it is sufficient to
communicate one "layer" of data between neighboring
processes/processors. In order to minimize the data volume which we
need to communicate, it makes sense that each processor deals with
contiguous data. In this fashion the "boundary" with neighboring
chunks is minimized. Furthermore, it is useful to define "ghost
cells" at internal boundaries. These ghost cells duplicate the
required additional data layer such that all differencing stencils
can be evaluated locally. The communication then consists in
up-dating the value of those ghost cells only.
- This is a 2D case, should i decompose the data in both
directions?
Good question. The answer is: yes. However, in this example we will
start with 1D decomposition for simplicity. Let us decompose the
second index only.
So, here comes the first task:
- write a routine which decomposes an integer number n into
nproc parts as evenly as possible. The routine has two
input arguments: scalar integers n, nproc; it has two
output arguments: two integer arrays of size nproc, containing -
respectively - the first and last index held by a processor 0<=myrank<=nproc-1.
Note: if the pointer arrays are called jbeg, jend, then
the local arrays will hold the index range jbeg(myrank)-1:jend(myrank)+1 because of the two "ghost
cells". Their dimension will then be jend(myrank)-jbeg(myrank)+2!
So, here comes the second task:
- Modify the sequential version of the jacobi program such that
- after the usual MPI initialization phase - every processor
computes these pointers. Dimension the locally held arrays
correctly. Attention: do you know how much data each processor
receives at compile time exactly? We suppose that nproc and the
system size n, m is known when compiling. If you do not
want to dynamically allocate arrays, a small over-dimensioning is
necessary.
Go ahead and modify the assignment of boundary conditions, the
jacobi step and the rest of the program such that you operate only
on the local bounds of the data.
The result should be a nearly-functional Jacobi program; the only
missing part now is the communication step which needs to be performed
after each iteration. An example for a driver routine is here:
So, here comes the third task:
- Write a communication subprogram which takes care of the
exchange of data between neighboring processors. Input arguments:
the local array, the pointers, the rank of the processor, the
communicator, the size of the communicator. Output arguments: the
array containing updated ghost-cell entries.
Things to keep in mind:
- the communication is pair-wise between neighboring
processes; each pair exchanges two lines of ghost-cell data; in
general each processor has two neighbors, which makes for 4
communication operations per processor - 2 sends and 2 receives.
- the available communication methods are either MPI_SEND and MPI_RECV or MPI_ISEND and MPI_IRECV or MPI_SENDRECV (which we have not treated
before). Watch the order!
- the "end processors" do not have 2 neighbors!
- ...
Possible solutions (of different quality!) are contained in the
following link:
Some hints for debugging the program:
- Did you forget one of the "five essentials" of an MPI
program?
include "mpif.h"
call MPI_INIT(ierr)
call call MPI_COMM_RANK(MPI_COMM_WORLD,myrank,ierr)
call MPI_COMM_SIZE(MPI_COMM_WORLD,nprocs,ierr)
call MPI_FINALIZE(ierr)
- Did you forget to declare the status array wherever you call
MPI_RECV?
integer istat(MPI_STATUS_SIZE)
- Did you correctly declare the local arrays and use the
pointers jbeg, jend consistently for transferring local
to global indices?
- A common mistake is forgetting that the processor rank varies
between 0 and nprocs-1 (and NOT 1 to nprocs).
- Include write statements. Most problems are due to the
fact that some information is not known to all processors or mixed
up. You will see many bugs immediately.
- Run parts of the code; the full code is probably more difficult
to debug.
- Check iterative convergence & convergence with number of modes
(should be second order).
- Plot the result & compare to exact solution; often you will see
where in the domain things do not work. How do you write a
distributed array to a single file? The practical way is to let one
"i/o master" process (e.g. myrank=0) receive and write data
to file, while all oters send their data to this master. Attention:
does the received data always fit into the master's local buffer?
Finally, we want to know which is the most efficient method of
communication (supposing that the rest of the code will be more or
less equivalent for all). You can obtain timing information by using
the MPI_WTIME function as follows:
t0=MPI_WTIME()
some code
t1=MPI_WTIME()
if(myrank.eq.0)write(*,*)'elapsed time: ',t2-t1
which will give the elapsed time in seconds. Strictly, the maximum
over all elapsed times should be computed, using e.g. MPI_REDUCE. Also: for timing purposes you should not run all
virtual processes on the master node, but
instead use the available ressources. If you simply execute
mpirun -np <no. of procs> <executable>
the script mpirun will decide where your processes are sent. You
can also force the selection by:
mpirun -np <no. of procs> -machinefile <file> <executable>
where <file> is the name of a file containing a
list of nodes (one per line) where processes should go, e.g.:
lince01.ciemat.es
lince02.ciemat.es
lince03.ciemat.es
lince04.ciemat.es
sends processes (in order) to nodes 01, 02, 03, 04. You can repeat
nodes in this file with the result that more than one process is
executed on that node.
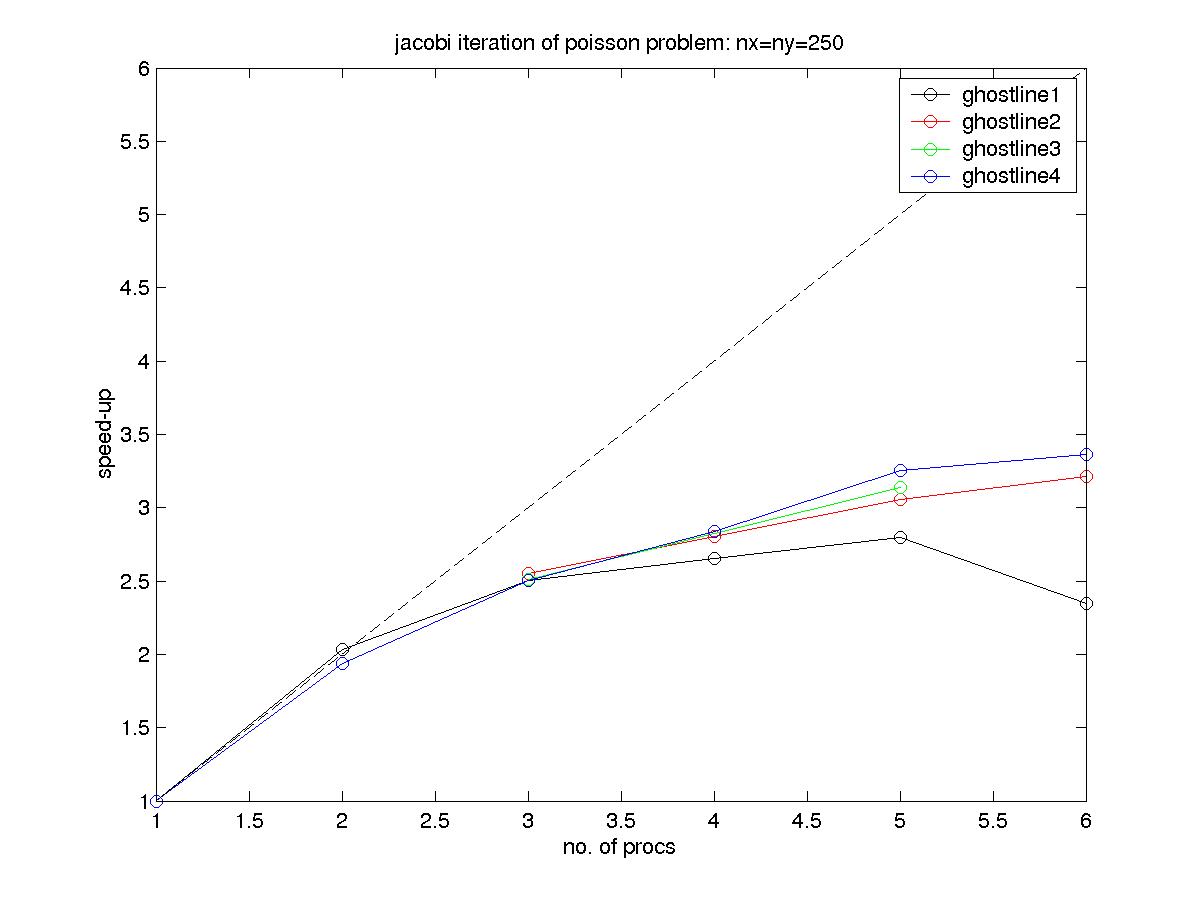
Results of timing the example solution - using the 4 different suplied
communication routines - on fenix.ciemat.es are plotted in this
graph (eps) (jpg)(tiff). You can
see that the ordering of the communication makes a difference! By the
way, relative parallel speed-up is measured as follows:
S=T1/Tp
where T1 is the execution time using one processor and Tp the
corresponding time for p processors. The other useful measure of
performance is parallel efficiency, computed as follows:
E=T1/(p*Tp)
Here, again all necessary routines to run the sequential and parallel
program are available for convenience:
markus.uhlmann AT ifh.uka.de



{kind=link}